
The lab’s research interests include
- computational epigenomics
- bioinformatics
- applied statistical learning
- chromatin biology
- single cell technology
- developmental and disease biology
Modeling Epigenetic Regulation of Cell Identity

More than 200 canonical cell types have been identified in different tissues of the human body. Although these cell types carry largely identical DNA sequences, regulatory mechanisms give rise to an abundance of cell phenotypes exhibiting markedly different degrees of specialization and differentiation. Our group is interested in computational approaches that can identify these regulatory mechanisms.
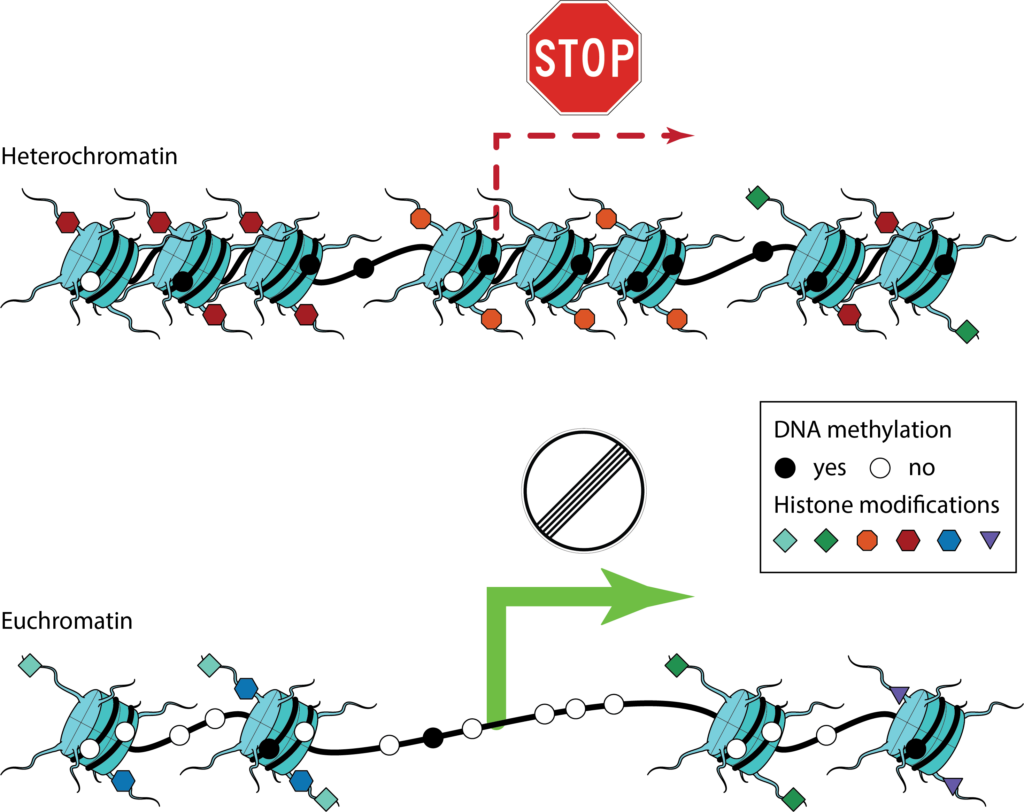
These regulatory mechanisms are to a large extent governed by the packaging and indexing of the DNA inside the cell’s nucleus. The entirety of DNA and associated proteins is called chromatin. It is compacted in a hierarchy of organizational structures and its packing density is associated with molecular factors, such as DNA-binding proteins, chromatin remodeling complexes as well as different, epigenetic modifications to the DNA or chromatin-associated proteins. Chromatin structure and associated epigenetic features have profound influence on how DNA acts as a physical molecule and thus play an essential role in governing gene regulation.
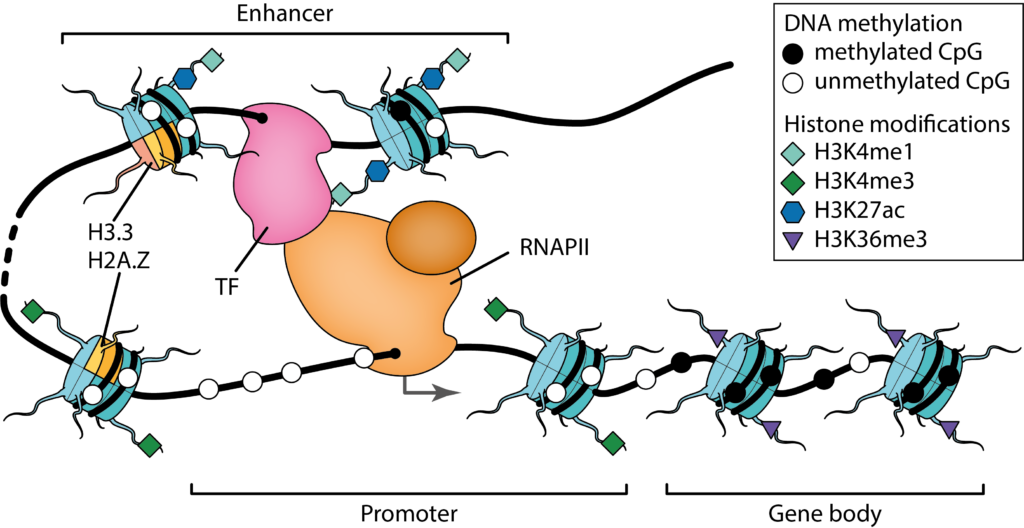
Our research focused on the statistical modeling of cell regulatory patterns. We are interested in inferring accurate models which can be interpreted from a molecular point of view and whose implied biological relationships can be evaluated experimentally. Two central questions of genome biology guide our research:
- Which epigenetic signatures contribute to establishing cell identity?
- Which regulatory interactions are involved in changing cell identity?
Our group develops and applies computational methods for the integrative analysis of epigenomic data by modeling the interplay between DNA sequence, DNA packaging (chromatin structure), spatial organization of the genome, regulatory enzymes and RNA molecules, and gene expression. This work involves the integration of large, heterogeneous datasets comprising epigenome-wide quantifications of different regulatory features in thousands of biological samples or millions of single-cells.
In order to facilitate reproducible research and method adoption, we are emphasizing the development of user-friendly software tools which implement the developed methods. These tools can be employed by biologists and bioinformaticians who can generate their own hypothesis through the analysis of reference and new data.
In short, we are interested in developing a computational framework of models that provide a quantitative definition of cell state and that describe the regulatory dynamics involved in developmental systems as well as in disease.